本編ではシングルセル解析初心者の方がテストデータを用いて、 一通りのデータ解析が行えるようになることを目的としています。
ここでは、トランスクリプトーム解析としてscRNA-seq、 エピゲノム解析としてscATAC-seq解析の方法を説明します。
リンク:シングルセル解析の必要性
リンク:シングルセル解析でできること・問題点
リンク:シングルセル解析の様々なプラットフォーム
ここでは、10x Genomics社のChromiumプラットフォームを用いた 配列データの解析方法について説明します。
Cell Rangerによる処理は多くのコンピュータ資源を必要としますので、高性能計算機の使用をお勧めします。 なおLoupeによる結果の閲覧は、通常のPCで多くの場合問題ありません。
この文書にある処理は東京大学医科学研究所ヒトゲノム解析センターのスーパーコンピュータ(HGC)で動作を確認しています。 HGCをご利用になる場合は、以下のメモリ使用指定でqloginしてください。
$ qlogin -l s_vmem=16G,mem_req=16G
Rをご使用する場合は、R/3.6以上のバージョンが必要です。 HGCをご利用の場合は、以下のように使用するRを指定してください。
$ module load R/3.6
リンク:シングルセル解析の様々なプラットフォーム
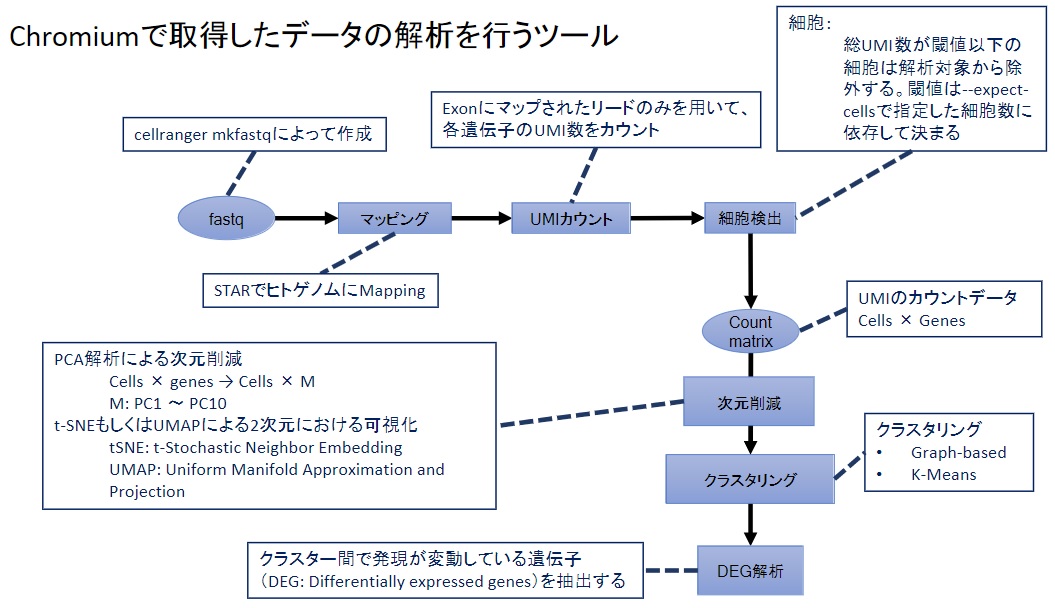
Chromiumを使ったscRNA-seqデータ解析
ここではマウス肺正常細胞のncRNA-seqデータと、解析ツールとしては10x Genomics社のCell Rangerを用いて、一連のパイプラインを処理してみます。
以下の操作について説明します。
- データの準備・ソフトウェアの確認
- Cell Rangerによる解析
- Cell Ranger実行結果の確認
- Loupe Cell Browserを用いたデータの確認
Cell Rangerとは
詳しくは以下 (英文)
https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger
データの準備・ソフトウェアの確認
- cellranger-4.0.0.tar.gz
- refdata-gex-mm10-2020-A.tar.gz
$ mkdir ~/tools/ #Cell Rangerをインストールするためのディレクトリを作成
$ cd ~/tools/ #ディレクトリ内に移動
$ #Cell Rangerのダウンロード
$ curl -o cellranger-4.0.0.tar.gz "https://cf.10xgenomics.com/releases/cell-exp/cellranger-4.0.0.tar.gz?Expires=1600871012&Policy=YYYYY&Signature=XXXXX"
$ tar -xzvf cellranger-4.0.0.tar.gz #解凍・展開
$ mkdir ~/tools/bin/ #ツールの実行ファイルを置くディレクトリを作成しておきます。
$ ln -s ~/tools/cellranger-4.0.0/cellranger ~/tools/bin/
$ #Referenceデータのダウンロード
$ mkdir ~/data/ #Cell Ranger用のリファレンスデータを設置するディレクトリを作成
$ cd ~/data/ #ディレクトリ内に移動
$ #Referenceデータセットをダウンロード
$ curl -O https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-mm10-2020-A.tar.gz
$ tar -xzvf refdata-gex-mm10-2020-A.tar.gz #ダウンロードしたファイルを解凍・展開
$ #Cell Rangerの動作確認
$ #~/tools/bin/にパスを通します
$ export PATH=~/tools/bin:$PATH
$ cellranger
cellranger 4.0.0
Process 10x Genomics Gene Expression, Feature Barcode, and Immune Profiling data
USAGE:
cellranger <SUBCOMMAND>
FLAGS:
-h, --help Prints help information
-V, --version Prints version information
SUBCOMMANDS:
count Count gene expression and feature barcoding reads from a single sample and GEM well
vdj Assembles single-cell VDJ receptor sequences from 10x Immune Profiling libraries
aggr Aggregate data from multiple Cell Ranger runs
reanalyze Re-run secondary analysis (dimensionality reduction, clustering, etc)
targeted-compare Analyze targeted enrichment performance by comparing a targeted sample to its cognate parent
WTA sample (used as input for targeted gene expression)
targeted-depth Estimate targeted read depth values (mean reads per cell) for a specified input parent WTA
sample and a target panel CSV file
mkvdjref Prepare a reference for use with Cell Ranger VDJ
mkfastq Run Illumina demultiplexer on sample sheets that contain 10x-specific sample index sets
testrun Execute the 'count' pipeline on a small test dataset
mat2csv Convert a gene count matrix to CSV format
mkgtf Prepare a GTF file for use as a 10x transcriptome reference
mkref Prepare a reference for use with 10x analysis software. Requires a GTF and FASTA
upload Upload a summary of an analysis pipeline job to 10x Genomics support
sitecheck Collect Linux system configuration information
help Prints this message or the help of the given subcommand(s)
$
テストデータの準備
$ #マウス肺細胞 Chromium scRNA-seq配列テストデータセットの準備
$ cd ~/data/ #データをダウンロードするディレクトリに移動
$ wget https://kero.hgc.jp/tutorials/learning/data/10X_RNAv3_Lung.tar
$ tar xvf 10X_RNAv3_Lung.tar
Cell Rangerによる解析
$ #適当な作業ディレクトリを作成し移動
$ mkdir ~/analysis_scRNA
$ cd ~/analysis_scRNA
$ #Cell Ranger count(アラインメント・カウント・各種解析(PCA, tSNE/UMAP, クラスタリング, 発現変動遺伝子(DEG)解析)コマンド)を実行します。
$ #引数の意味は以下の通りです
$ #--id: 任意の解析のID
$ #--transcriptome: 10x Genomics社のHPからダウンロードしたreferenceデータセットを展開したディレクトリを指定
$ #--fastqs: chromiumの出力したfastqファイルの入ったディレクトリを指定
$ #--expect-cells: 予想されるシングルセル数(=10000)
$ #--localcores: 解析に使うCPUコア数(ご自身の環境に合わせてなるべく大きな値にしてください)
$ #--localmem: 使用メモリ量(GB)(ご自身の環境に合わせてなるべく大きな値にしてください)
$ cellranger count --id=emm_rnaseq --transcriptome=../data/refdata-gex-mm10-2020-A/ --fastqs=../data/10X_RNAv3_Lung/ --expect-cells=10000 --localcores=8 --localmem=90
$ #...解析完了までお待ちください(上のコマンドはhgcのスーパーコンピュータで処理した場合ですが、6~7時間を要しました)
Cell Ranger 実行結果の確認
$ cd ~/analysis_scRNA/emm_rnaseq/
$ ls
SC_RNA_COUNTER_CS _filelist _invocation _log _perf _tags _uuid _versions outs
_cmdline _finalstate _jobmode _mrosource _sitecheck _timestamp _vdrkill emm_rnaseq.mri.tgz
$ #outsに解析結果ファイルが出力されます
$ cd outs
$ ls
analysis filtered_feature_bc_matrix.h5 possorted_genome_bam.bam raw_feature_bc_matrix.h5
cloupe.cloupe metrics_summary.csv possorted_genome_bam.bam.bai web_summary.html
filtered_feature_bc_matrix molecule_info.h5 raw_feature_bc_matrix
以下のような結果ファイルが出力されます。
| File Name | Description |
|---|---|
| web_summary.html | HTML形式のサマリー。 |
| metrics_summary.csv | CSV形式のサマリー。 |
| possorted_genome_bam.bam | BAMファイル、IGVで閲覧可能。 |
| possorted_genome_bam.bam.bai | BAIファイル。 |
| filtered_feature_bc_matrix | 細胞ごとの各遺伝子のUMI数のデータ。filter(UMI数の閾値)をパスした細胞のデータのみ。 |
| filtered_feature_bc_matrix.h5 | HDF5形式。細胞ごとの各遺伝子のUMI数のデータ。filterをパスした細胞のみ。 |
| raw_feature_bc_matrix | 細胞ごとの各遺伝子のUMI数のデータ。Filterではじかれたデータも含む。 |
| raw_feature_bc_matrix.h5 | HDF5形式。細胞ごとの各遺伝子のUMI数のデータ。Filterではじかれたデータも含む。 |
| analysis | DEGや、クラスタリング結果の情報を含むディレクトリ。 |
| molecule_info.h5 | Cell Rangerの再解析に使用する。 |
| cloupe.cloupe | Loupe Cell Browser用のファイル。 |
結果ファイル web_summary.html
Summaryタグ
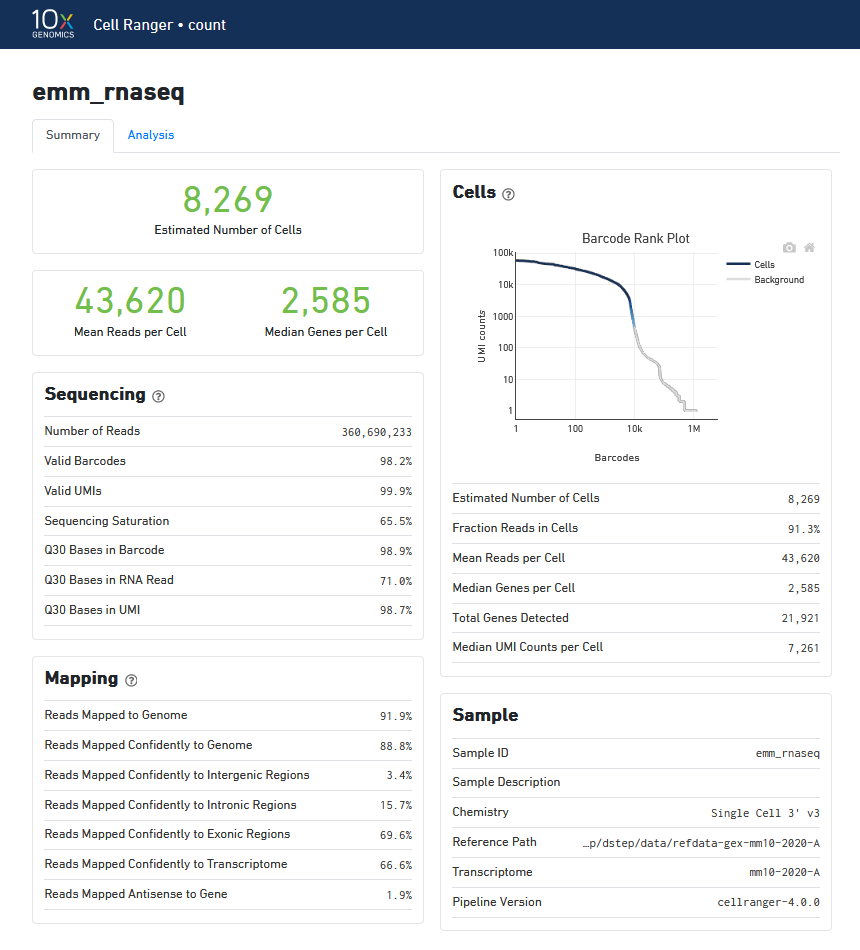
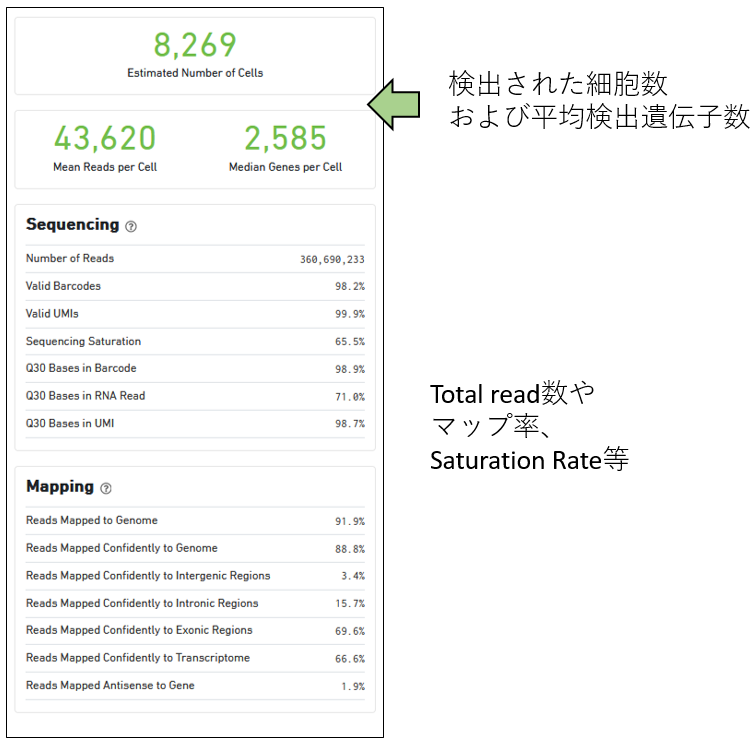
|
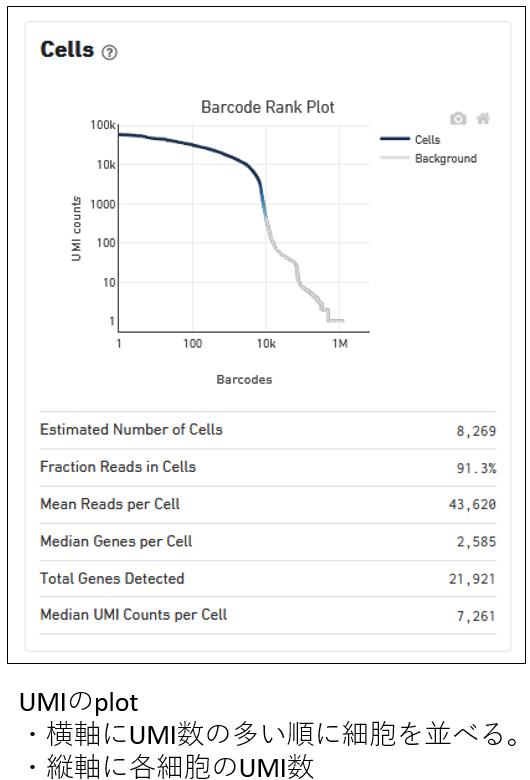
|
Analysisタグ
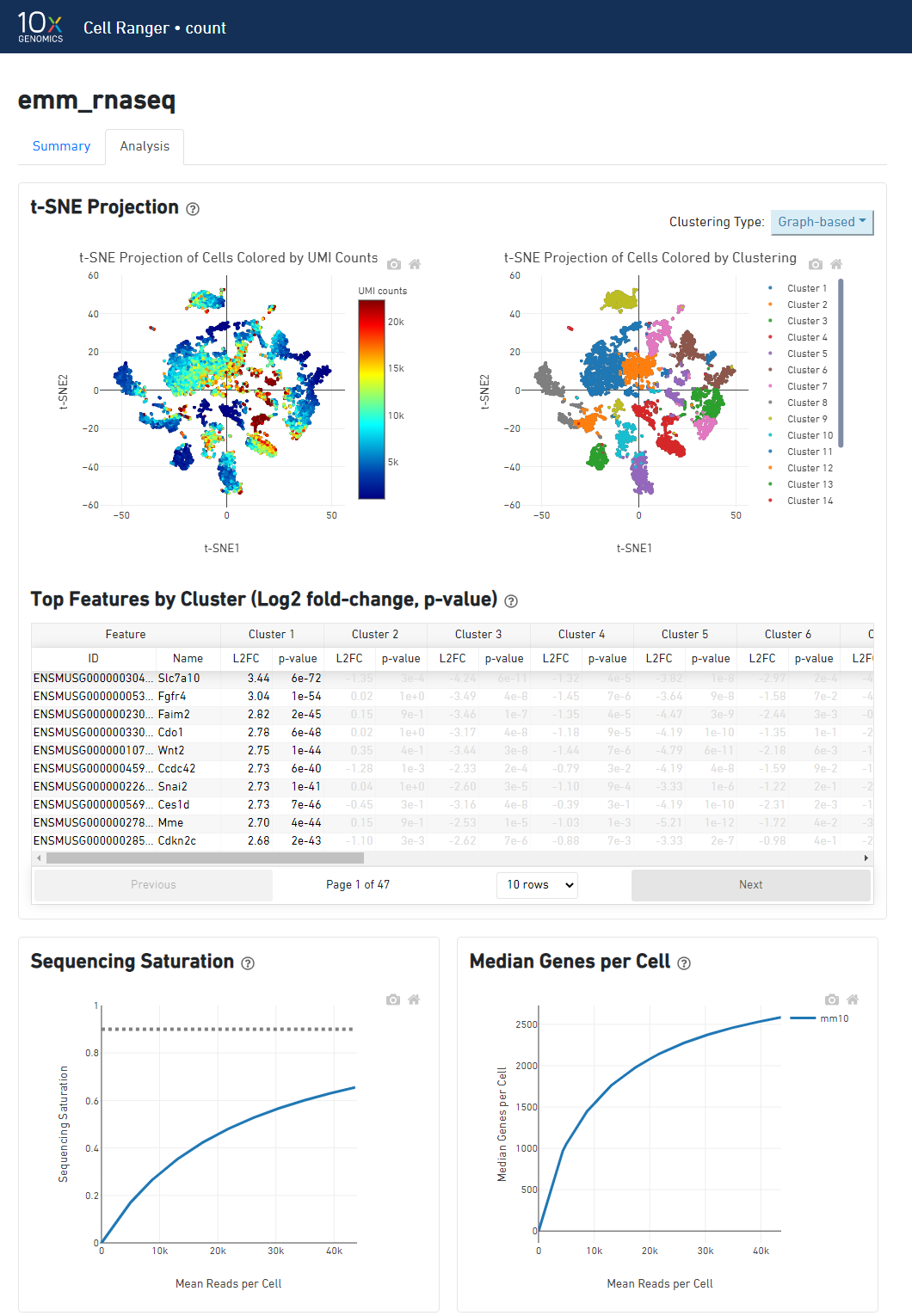
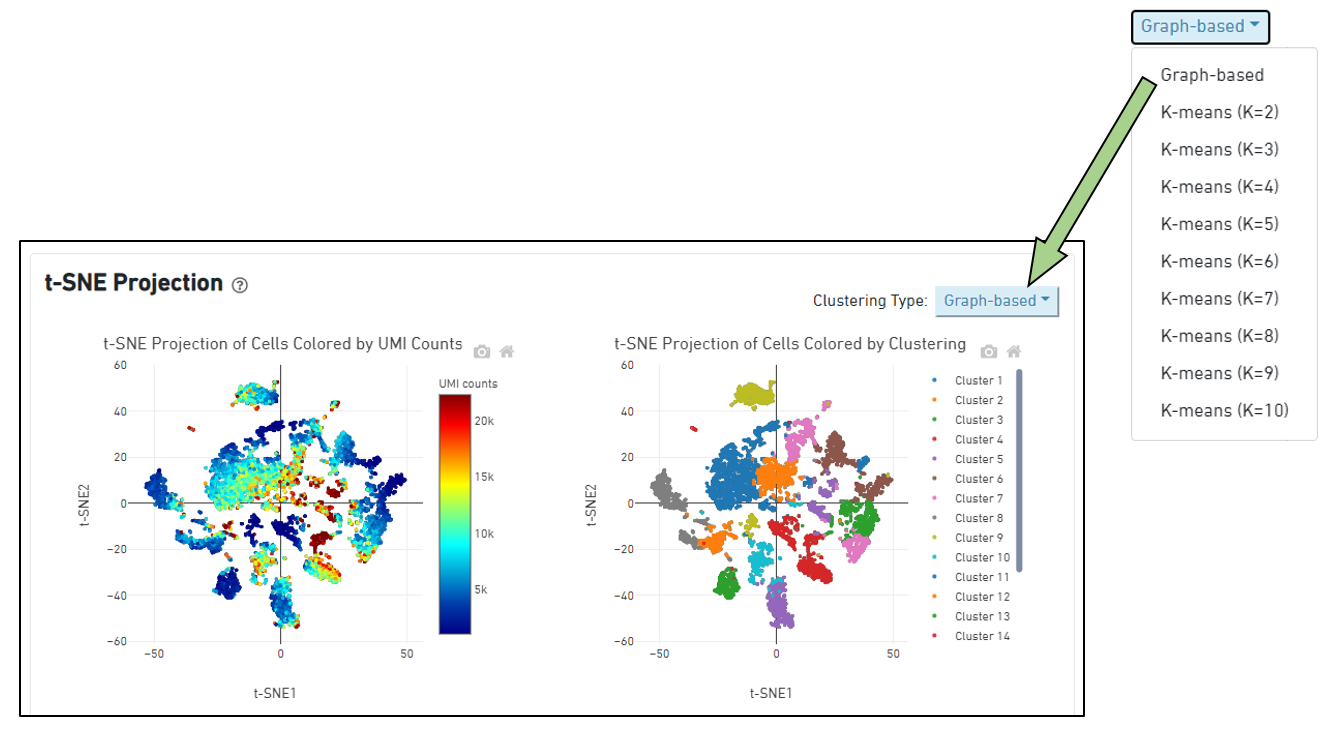
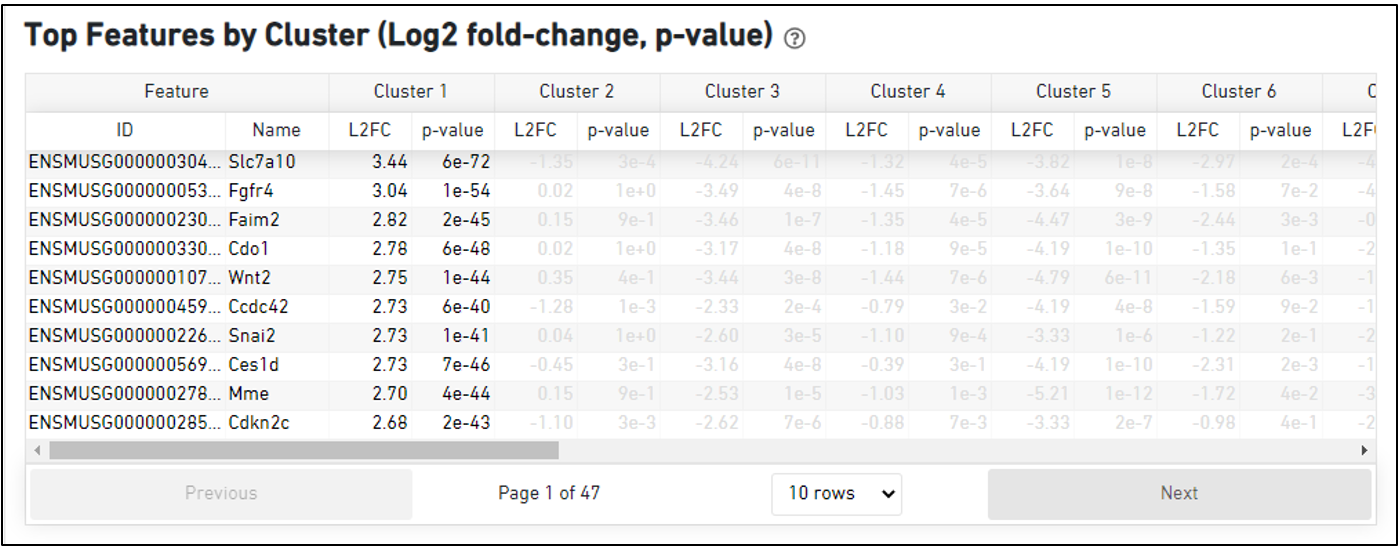

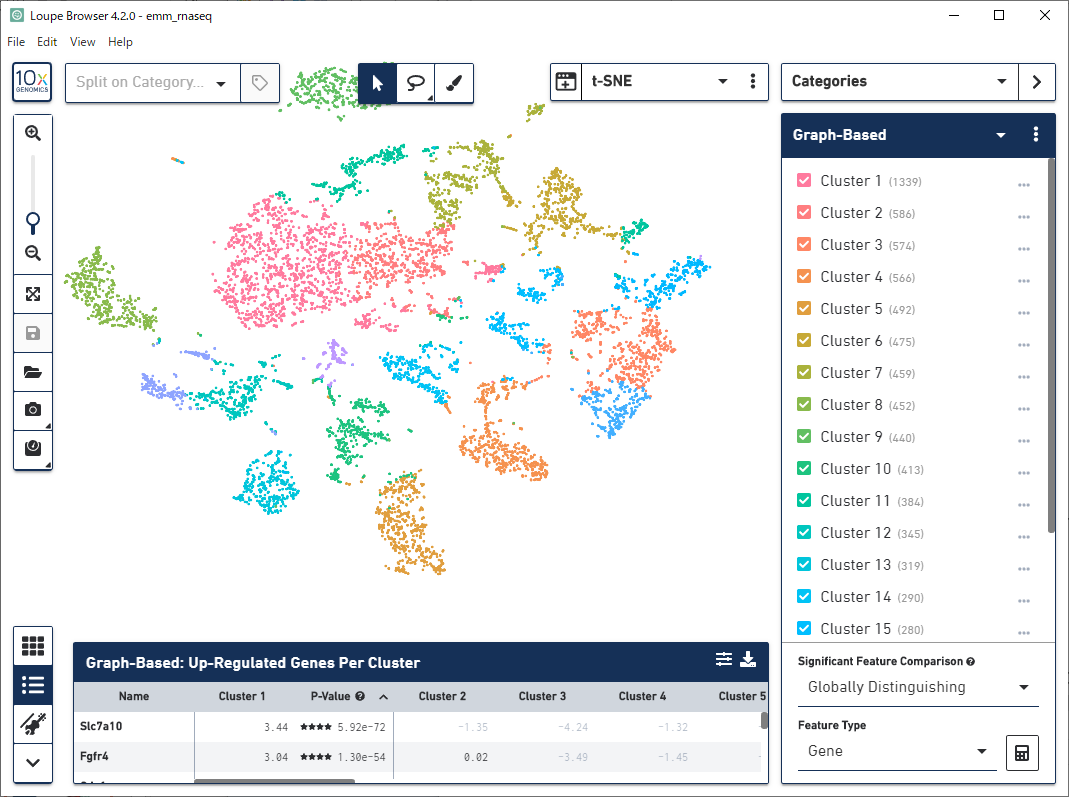
結果ファイル cloupe.cloupe
この結果ファイルから以下のことができます。
- Loupe Cell Browserを使用して、グラフィカルにCell Rangerの結果を可視化することができる。
- 特定細胞集団の抽出や、任意で作成したクラスター間のDEGの検出を行うことができる。
- 詳細な使い方は次のチュートリアルを参考に。What is Loupe Browser?
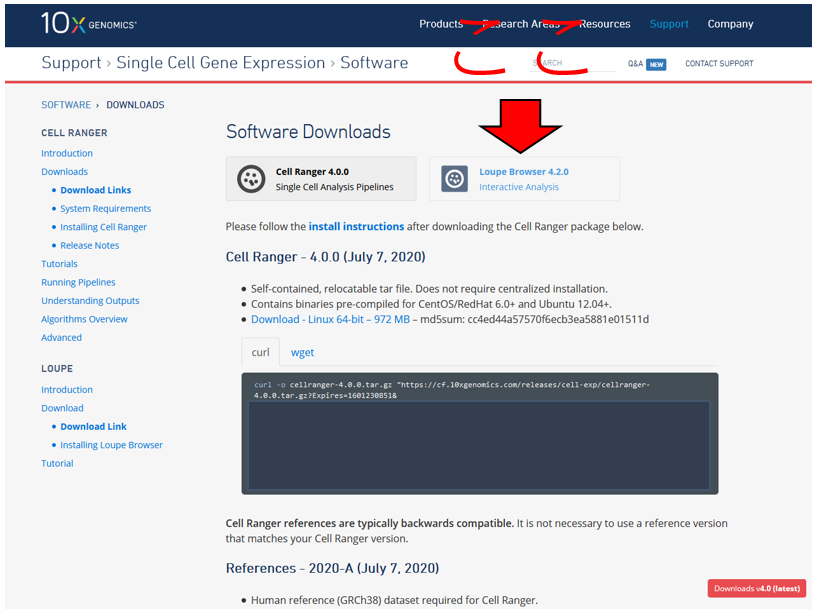
お使いのクライアントPCにLoupe Cell Browserを以下のURLからダウンロード・インストールしましょう。
https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest
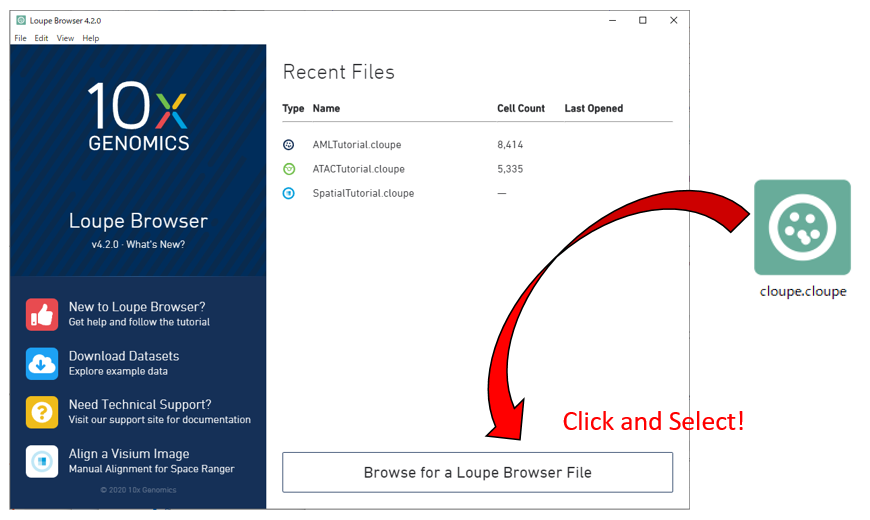
cloupe.cloupeファイルをPCにダウンロードしてLoupe Cell Browserで開きましょう。
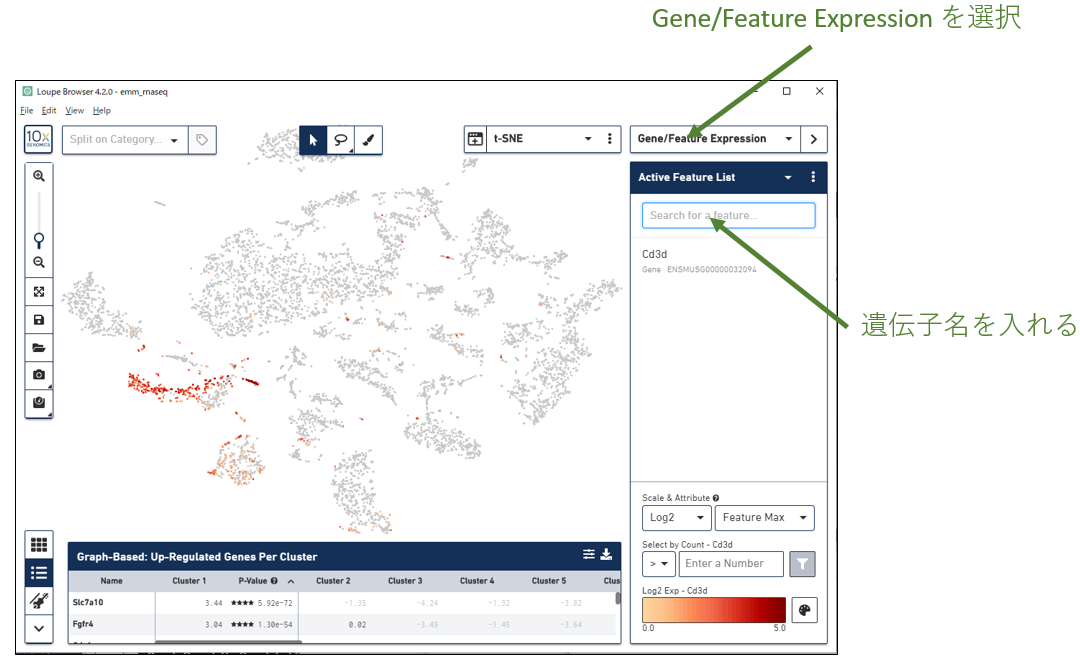
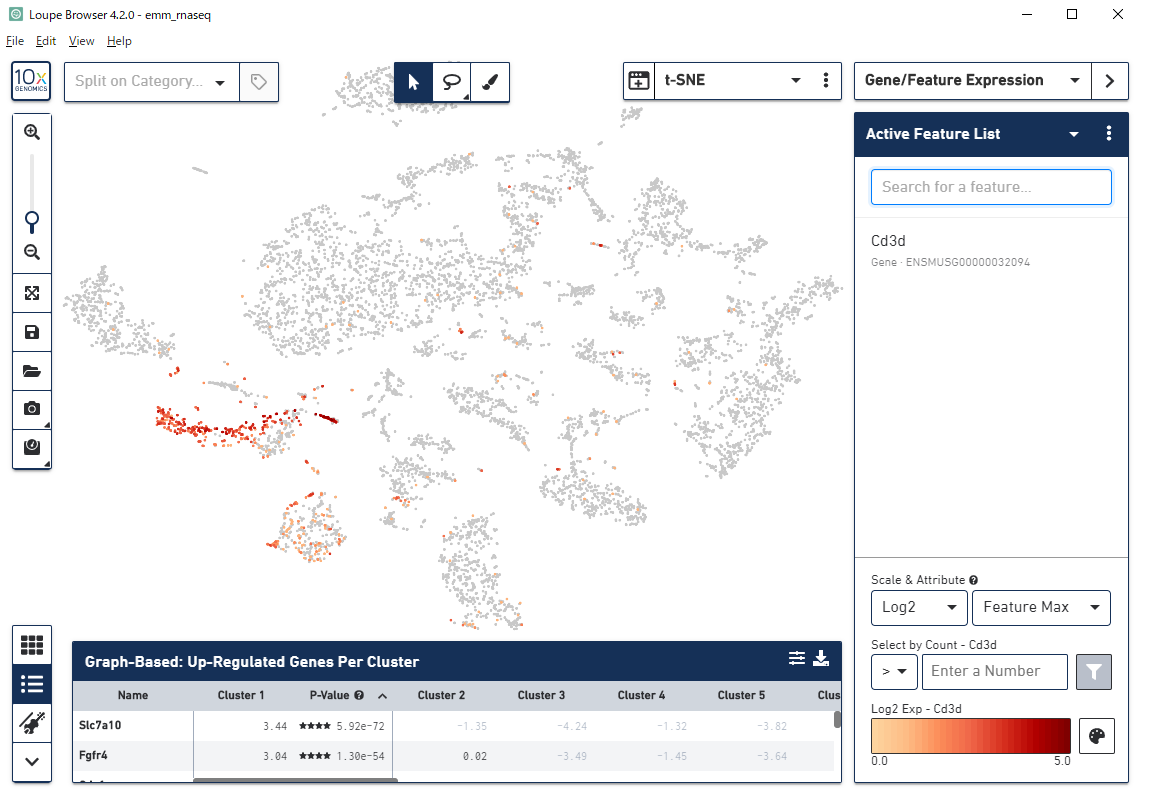
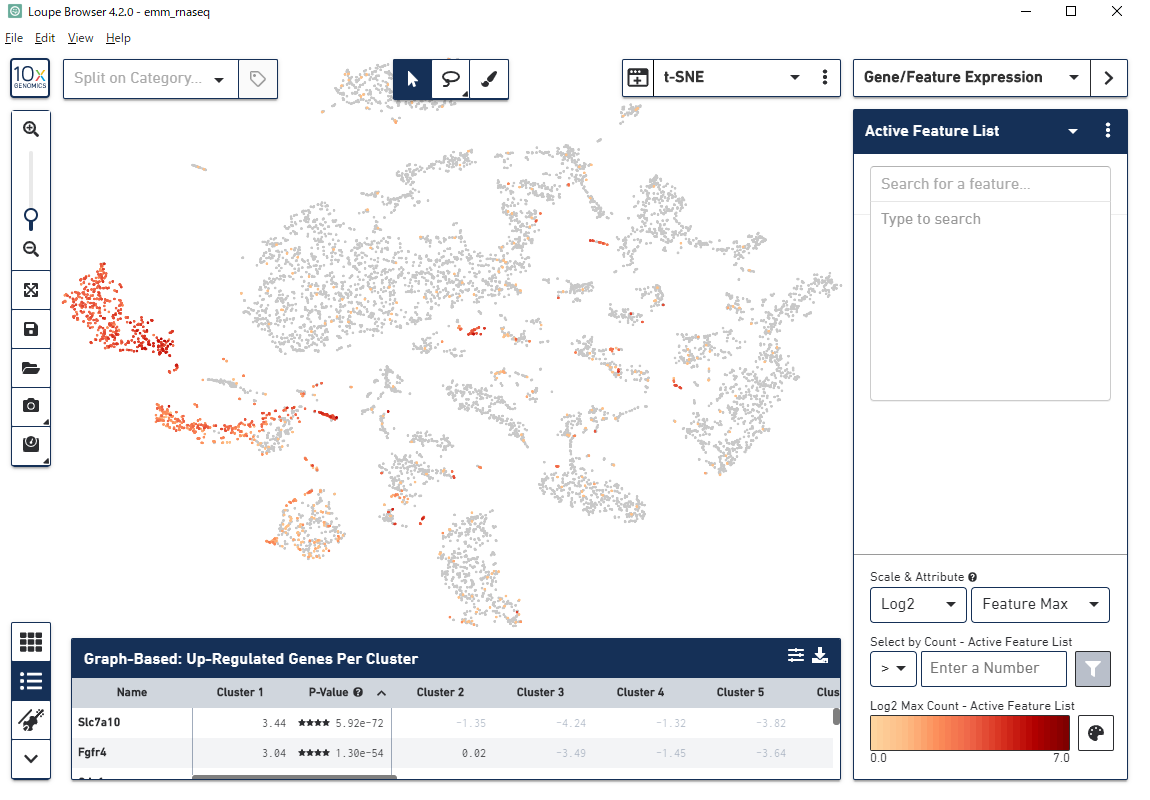
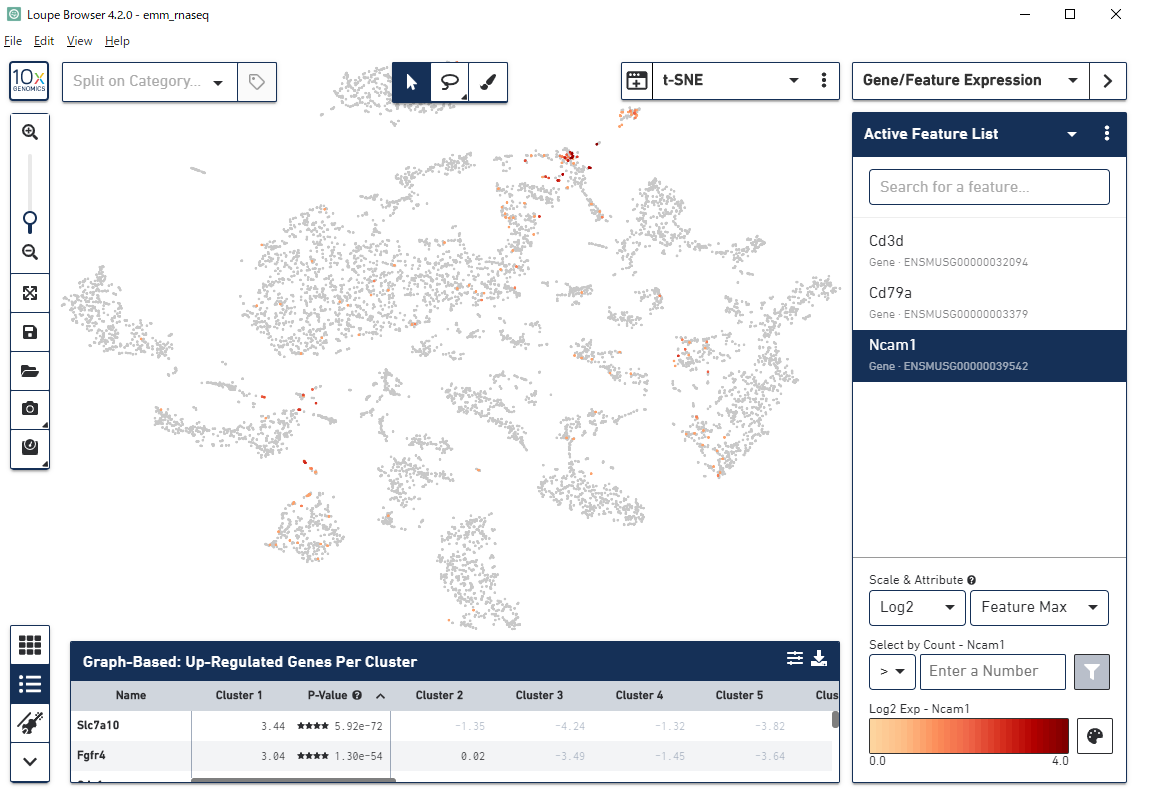
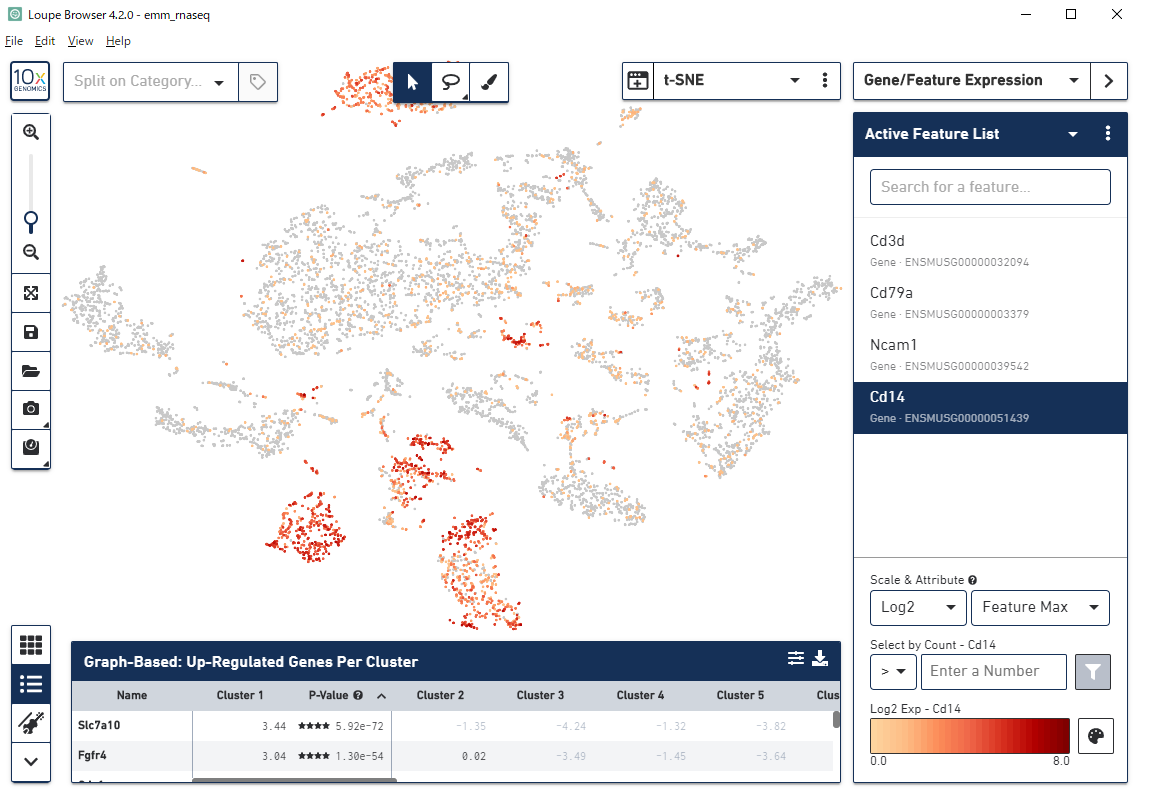
アノテーション・細胞種同定
マーカー遺伝子の発現量確認
|
|
細胞種同定
|
|
|
|

|

|
Chromiumを使ったscATAC-seqデータ解析
ここではマウス肺正常細胞のncATAC-seqデータと、解析ツールとしては10x Genomics社のCell Ranger ATACを用いて、一連のパイプラインを処理してみます。
以下の操作について説明します。
- データの準備・ソフトウェア確認
- Cell Ranger ATACによる解析
- Cell Ranger ATAC 実行結果の確認
- Loupe Cell Browserを用いたデータの確認
Cell Ranger ATACとは
- Alignment: アダプタートリミングをして、BWA-MEMでマッピング
- Duplicate Marking: PCR duplicateリードをチェック
- Peak Calling: ATAC-seqピーク(オープンクロマチン領域)を検出
- Cell Calling: リードがピーク領域にオーバーラップしているリードが一定以上ある細胞を採用
- Peak-Barcode Matrix: ピークと細胞のマトリックスを作成
- Dimensionality Reduction, Clustering, t-SNE Projection: 次元削減(LSA)→ クラスタリング→ 2D可視化
- Peak Annotation: 近傍遺伝子を紐づけ、プロモーター・遺伝子領域のカウント、転写因子結合部位の同定
- Transcription Factor Motif Enrichment Analysis: 各細胞のピークに有意に見出される転写因子モチーフの解析
- Differential Accessibility Analysis: クラスター間における転写因子結合モチーフの違いを解析
詳しくは以下
https://support.10xgenomics.com/single-cell-atac/software/pipelines/latest/algorithms/overview
データの準備・ソフトウェアの確認
10x Genomics社のダウンロードのページより、 最新版のCell Ranger ATAC(1.2.0)とreferenceデータセット(mouse mm10)(2020/09/23執筆時)をダウンロードします。 ※メールアドレスの登録が必要となります。- cellranger-atac-1.2.0.tar.gz
- refdata-cellranger-atac-mm10-1.2.0.tar.gz
$ mkdir -p ~/tools/ #Cell Ranger ATACをインストールするためのディレクトリを作成(既に作成してある場合は不要)
$ cd ~/tools/ #ディレクトリ内に移動
$ #Cell Ranger ATACのダウンロード
$ curl -o cellranger-atac-1.2.0.tar.gz "https://cf.10xgenomics.com/releases/cell-atac/cellranger-atac-1.2.0.tar.gz?Expires=WWWWW&Policy=ZZZZZ&Signature=YYYYY&Key-Pair-Id=XXXXX"
$ tar -xzvf cellranger-atac-1.2.0.tar.gz #解凍・展開
$ mkdir -p ~/tools/bin/ #ツールの実行ファイルを置くディレクトリを作成しておきます。(既に作成してある場合は不要)
$ ln -s ~/tools/cellranger-atac-1.2.0/cellranger-atac ~/tools/bin/
$ #Referenceデータのダウンロード
$ mkdir -p ~/data/ #Cell Ranger用のリファレンスデータを設置するディレクトリを作成(既に作成してある場合は不要)
$ cd ~/data/ #ディレクトリ内に移動
$ #Referenceデータセットをダウンロード
$ curl -O https://cf.10xgenomics.com/supp/cell-atac/refdata-cellranger-atac-mm10-1.2.0.tar.gz
$ tar -xzvf refdata-cellranger-atac-mm10-1.2.0.tar.gz #ダウンロードしたファイルを解凍・展開
$ #Cell Rangerの動作確認
$ #~/tools/bin/にパスを通します(既に通してある場合は不要)
$ export PATH=~/tools/bin:$PATH
$ cellranger-atac
cellranger-atac (1.2.0)
Copyright (c) 2019 10x Genomics, Inc. All rights reserved.
-------------------------------------------------------------------------------
Usage:
cellranger-atac mkfastq
cellranger-atac count
cellranger-atac aggr
cellranger-atac reanalyze
cellranger-atac mkref
cellranger-atac testrun
cellranger-atac upload
cellranger-atac sitecheck
$
テストデータの準備
$ #マウス肺細胞 Chromium scATAC-seq配列テストデータの準備
$ cd ~/data/ #データをダウンロードするディレクトリに移動
$ wget https://kero.hgc.jp/tutorials/learning/data/10X_ATAC_Lung.tar
$ tar xvf 10X_ATAC_Lung.tar
Cell Ranger ATACによる解析
$ #適当な作業ディレクトリを作成し移動
$ mkdir ~/analysis_scATAC
$ cd ~/analysis_scATAC
$ #cellranger-atac count(アライメント、ピークコール、解析(次元削減、クラスタリング等)を行うコマンド)を実行します。
$ #--id: 任意の解析のID
$ #--reference: 10x Genomics社のHPからダウンロードしたreferenceデータセットを展開したディレクトリを指定
$ #--fastqs: chromiumの出力したfastqファイルの入ったディレクトリを指定
$ #--localcores: 解析に使うCPUコア数(ご自身の環境に合わせて大きな値にしてください)
$ #--localmem: 使用メモリ量
$ cellranger-atac count --id=emm_atac --reference=../data/refdata-cellranger-atac-mm10-1.2.0/ --fastqs=../data/10X_ATAC_Lung/ --localcores=8 --localmem=90
$ #...解析完了までお待ちください(上のコマンドはhgcのスーパーコンピュータで処理した場合ですが、約1日を要しました)
Cell Ranger ATAC 実行結果の確認
$ cd ~/analysis_scATAC/pbmc_5k_atac
$ cd ~/analysis_scATAC/emm_atac/
$ ls
SC_ATAC_COUNTER_CS _jobmode _sitecheck _versions
_cmdline _log _tags emm_atac.mri.tgz
_filelist _mrosource _timestamp outs
_finalstate _perf _uuid
_invocation _perf._truncated_ _vdrkill
$
$ #outsに解析結果ファイルが出力されます
$ cd outs
$ ls
analysis fragments.tsv.gz raw_peak_bc_matrix
cloupe.cloupe fragments.tsv.gz.tbi raw_peak_bc_matrix.h5
filtered_peak_bc_matrix peak_annotation.tsv singlecell.csv
filtered_peak_bc_matrix.h5 peaks.bed summary.csv
filtered_tf_bc_matrix possorted_bam.bam summary.json
filtered_tf_bc_matrix.h5 possorted_bam.bam.bai web_summary.html
以下のような結果ファイルが出力されます。
| File Name | Description |
|---|---|
| web_summary.html | HTML形式のサマリー。 |
| summary.csv | CSV形式のサマリー。 |
| possorted_bam.bam | BAMファイル、IGVで閲覧可能。 |
| possorted_bam.bam.bai | BAIファイル。 |
| summary.json | JSON形式のサマリー。 |
| peaks.bed | ピーク領域。 |
| raw_peak_bc_matrix | 細胞ごとのPeakのデータ。 |
| raw_peak_bc_matrix.h5 | HDF5形式。細胞ごとのPeakデータ。 |
| filtered_gene_bc_matrices | 細胞ごとのPeakデータ。filter(閾値)をパスした細胞のデータのみ。 |
| filtered_gene_bc_matrices.h5 | HDF5形式。細胞ごとのPeakデータ。filterをパスした細胞のみ。 |
| fragments.tsv.gz | BED-likeの形式。Fragmentの情報。Duplicateは一つにまとめられている。 |
| fragments.tsv.gz.tbi | Fragmentのindexファイル。 |
| filtered_tf_bc_matrix | 細胞ごとの転写因子のデータ。 |
| filtered_tf_bc_matrix.h5 | HDF5形式。細胞ごとの転写因子のデータ。 |
| analysis | DEGや、クラスタリング結果の情報を含むディレクトリ。 |
| peak_annotation.tsv | Peak領域に遺伝子情報を紐づけたもの。 |
| cloupe.cloupe | Loupe Cell Browser用のファイル。 |
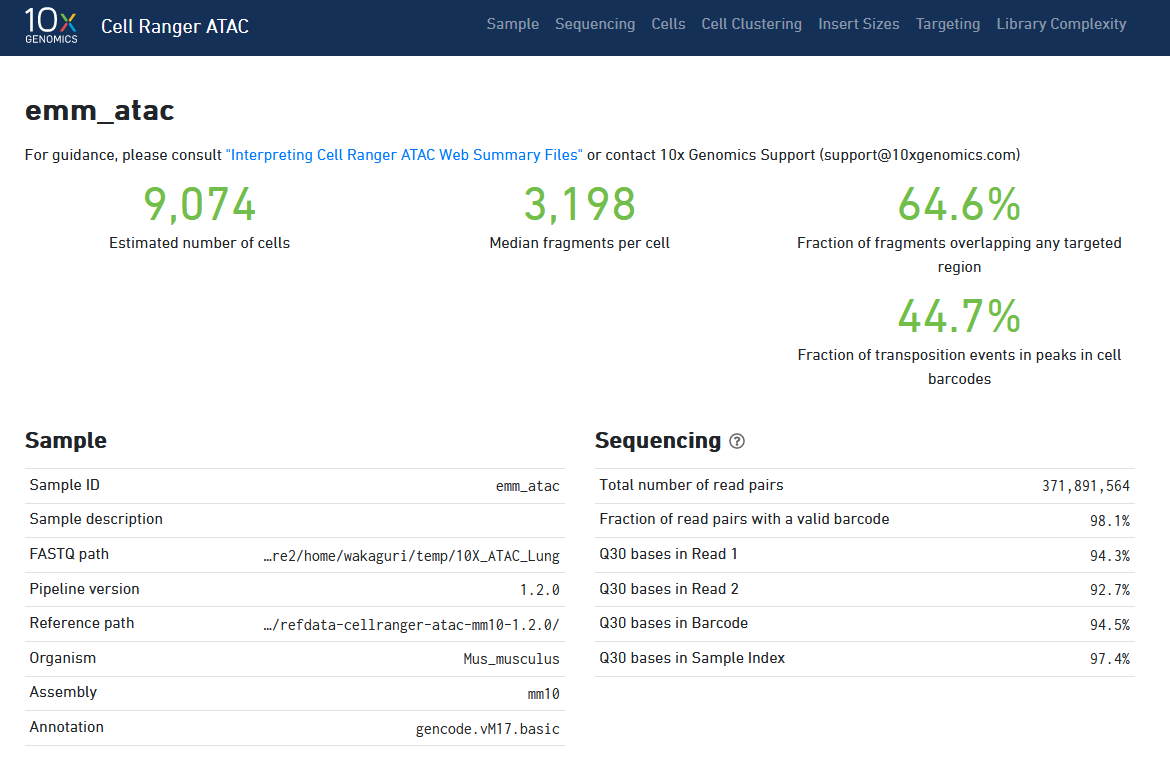
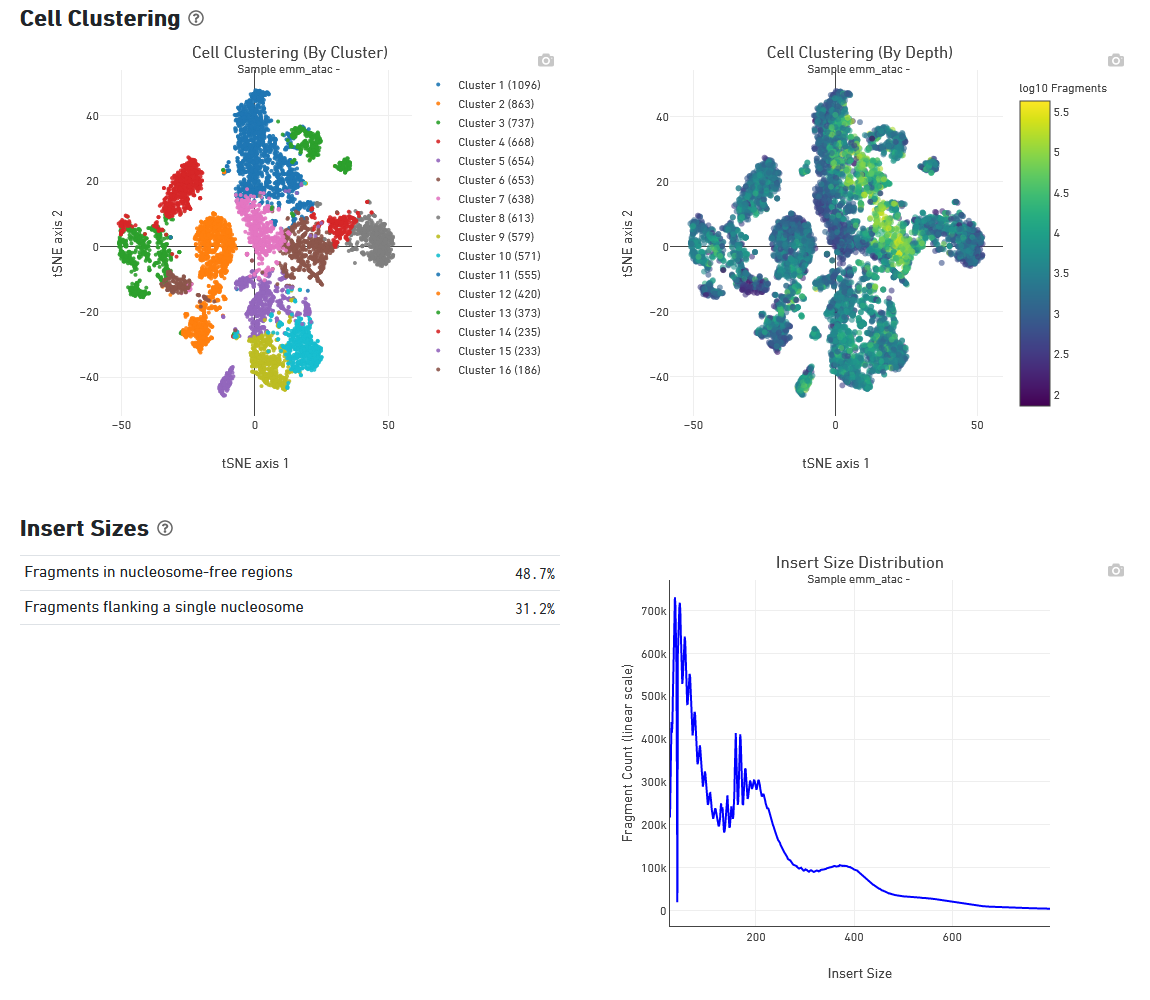
結果ファイル web_summary.html
「?」を押せば各項目の説明を見ることができます。


Loupe Cell Browserを用いた結果の吟味
まだお使いのクライアントPCにLoupe Cell Browserをダウンロード・インストールしてない方は、上記を参考にインストールしましょう。
結果ファイル cloupe.cloupe

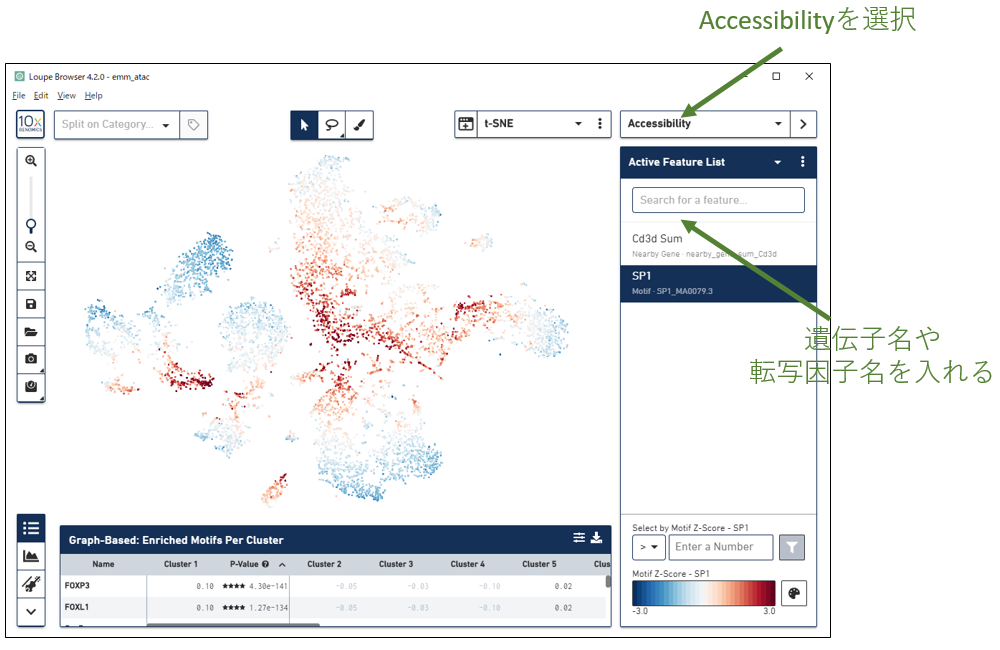
アノテーション・細胞種同定
マーカー遺伝子のAccesibility確認